Calculation Linkage Disequilibrium From Poolseq Data
Calculating linkage disequilibrium (LD) from Pool-seq data
[2019-12-06]
- Pool sequencing data does not contain individual genotype data and hence the linear combinations of alleles per genotype cannot be known. In other words haplotype information is lost.
- What we know are allele frequencies per pool
- We have linear allele combination information per read which is limited by the sequencing technology used, i.e. for the hugely popular and currently state-of-the-art short-read fluorescence-based sequencing technology by Illumina, this is limited to 150bp tp 300bp, and an impressive ~2Mbp using nanopore sequencing.
- Considering short-read sequencing, should we look for overlapping sequences (which can be very rare if they exist at all) to somehow piece-together a longer sequence. This approach will not work for deliberately sparse genome coverage approches such as genotyping-by-sequencing (GBS) and restriction site-associated DNA sequencing (RADseq).
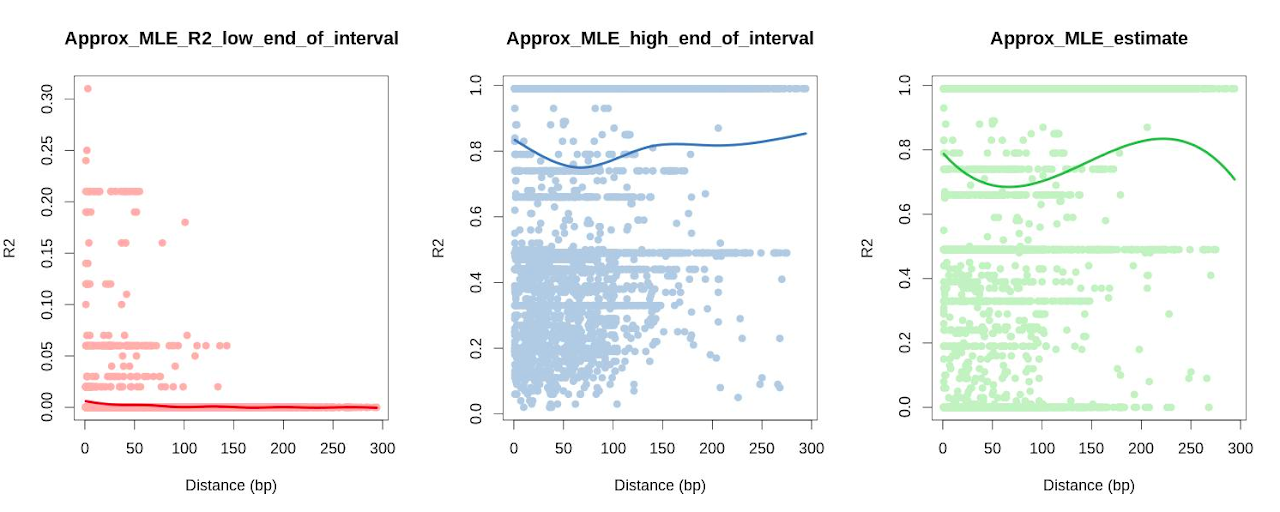
- Tests using LDx which does computes LD per sequencing read and therefore severely limited by maximum read length; shows that wthin this short range of 0-300 bp, the decay of LD is not yet evident (see scatterplots below).